
NeuralXC: a machine learning method to create accurate DFT density functionals
Machine learning accurate exchange and correlation functionals of the electronic density

Sebastian Dick and Marivi Fernandez-Serra
Density functional theory (DFT) is the standard tool to study the electronic structure of materials and condensed systems. While nowadays wave function based methods (inherently more accurate than DFT) are becoming more and more accesible (thanks in part to codes like pyscf) , the size of the systems they can simulate is still on the small side. DFT provides an optimal balance between accuracy and computational cost, making a first-principles description of complex and large systems possible.
To achieve this balance, DFT is mapped onto a mean-field single electron description within the Kohn-Sham (KS) approach. In KS-DFT, all the complexities of the many-body electron-electron interaction are reduced within a functional of the density. This functional consists of an exchange (X) and a correlation (C) part, the former capturing effects from Pauli-exchange, and the latter approximating correlations of electrons within the many-body wave function.
In this work by, led by Sebastian Dick we propose a pathway to construct fully machine-learned functionals that depend explicitly on the electronic density and implicitly on the atomic positions and are built on top of physically motivated functionals in a Delta-learning type approach.
Our method is an evolution of our recent work, in which we developed machine-learned correcting functionals (MLCF) to correct energies and forces by learning from the electron density.
Building on it, in this work, we show that it is possible to take the functional derivative of MLCFs thus creating semilocal ML KS density functionals that can be used in self-consistent calculations. We call this overall method NeuralXC.
We show that these functionals encode meaningful chemical information that extends beyond the training set, hence making the functionals transferable.

The picture above shows a summary of how our method works. In a nutshell, we had to identify the appropriate model for the problem. The model (which includes the neural network architecture and the descriptors) needs to satisfy a number of conditions:
Should be system and size independent. We want to create a real XC functional, valid for any system and not only for small molecules.
Should respect all physical symmetries.
Should permit the computation of functional derivatives.
Should be transferable.
We achieve this by a combination of rotationally invariant atom-centered density basis set descriptors and a set of permutationally invariant Behler-Parrinello networks to parametrize the energy functional.
Starting from the electron density in real-space, obtained with a converged DFT calculation, the projector maps this density to a set of descriptors. The symmetrizer creates rotationally invariant versions of these descriptors, which, after preprocessing (not depicted here), are passed through a Behler-Parrinello type neural network architecture. By using the same network for descriptors of a given atomic species, we ensure permutation invariance. Once the energy is obtained, its derivative can be back propagated using the chain rule to obtain the machine learned potential. This potential is added back to the baseline potential and the resulting potential can be used in subsequent self-consistent calculations.
In our paper we show that using NeuralXC, it is possible to create specialized func tionals that are highly accurate when used in systems sufficiently similar to their training data, while not degrading the overall accuracy of their baseline method (and in some cases improving it) when used far outside their training domain.
Another cornerstone of a successful ML model is its transferability, facilitating model creation itself. As NeualXC functionals generalize across chemical environments, the need to create a new reference dataset and re- train a new model for each system of interest decreases.
Using the method, we created a new XC functional for water, NXC-W01. This functional outperforms any other DFT functional on its description of water. Beyond reproducing pair-correlation functions close to experimental results, it is capable of treating bond-breaking, opening the path to studying proton transfer processes in liquid water at a highly accurate level.
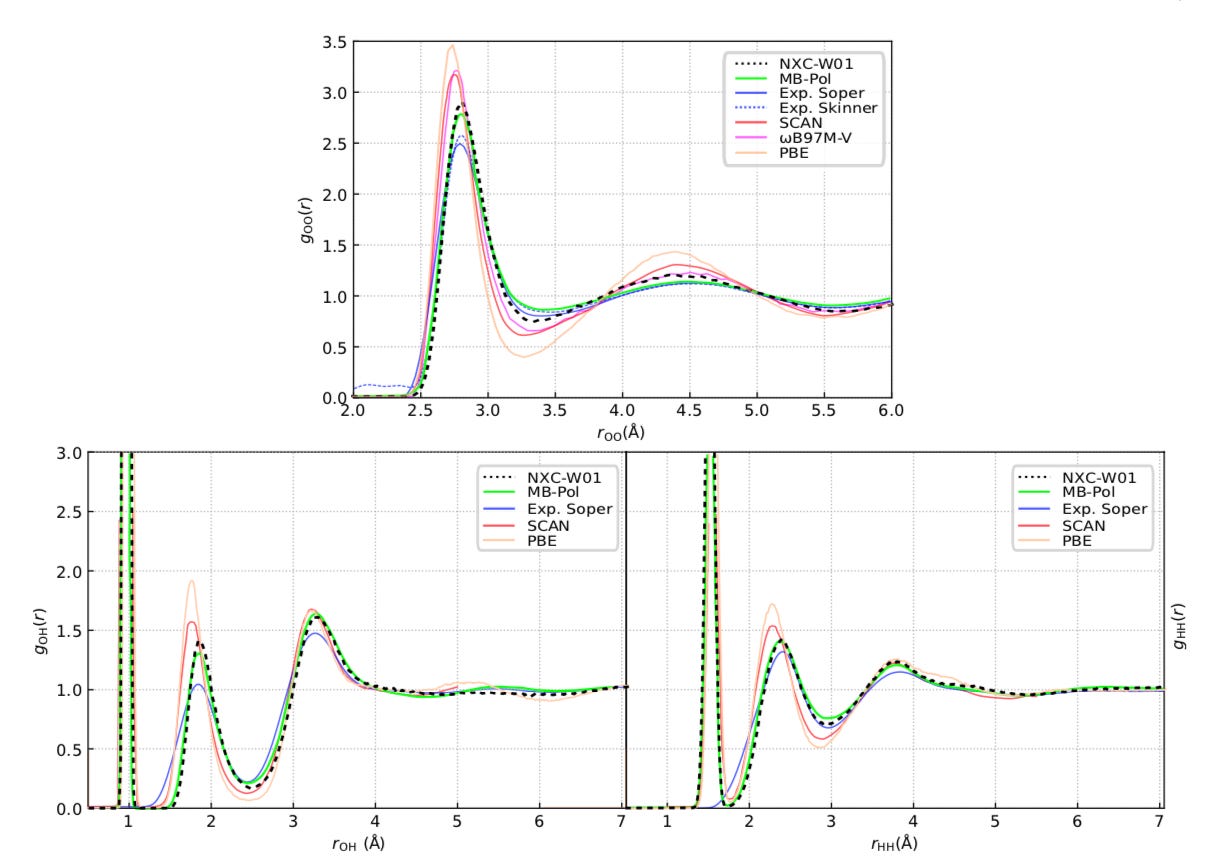

NeuralXC opens up a new path to developing exchange-correlation functionals for KS-DFT calculations. As our method only introduces a linearly scaling overhead to the underlying baseline functional, it is especially attractive for simulations of large systems for which explicitly correlated wave-function methods are still too expensive.